Capítulo 2
El pianista demente
El 15 de abril de 2003, los periódicos, los programas de televisión y las páginas web de todo el mundo dieron la noticia: el mapa del genoma humano se había completado.
Solo había un problemilla de nada: no era verdad. De hecho, había enormes lagunas en la secuencia.
No se trataba de otro caso en el que los medios de comunicación de masas estaban exagerando. Revistas científicas de gran prestigio, como Science o Nature, contaron lo mismo. Tampoco estaban los científicos exagerando su trabajo. La verdad era que, en esos días, la mayoría de los investigadores implicados en el proyecto, que duró trece años y costó mil millones de dólares, estaba de acuerdo en que era lo más cerca que podríamos estar, debido a la tecnología de la época, de identificar cada uno de los tres mil millones de pares de bases de nuestro ADN.
Las partes del genoma que faltaban, normalmente secciones solapadas de nucleótidos repetitivos, no se consideraron importantes. Estas eran partes del código de la vida que en otro tiempo se tachaban de «ADN basura», pero que ahora merecen algo más de respeto, aunque se siguen desestimando como «no codificante». Desde la perspectiva de los mejores científicos de la época, esas regiones eran poco más que fantasmas de genomas pasados, en su mayoría remanentes de virus oportunistas muertos que se integraron en el genoma hace cientos de miles de años. Lo que nos hace ser lo que somos, se creía por entonces, se había identificado en su mayoría, y ya contábamos con lo necesario para ampliar nuestro entendimiento de lo que nos hace humanos.
Sin embargo, algunas estimaciones indican que esa materia genética oscura asciende al 69 por ciento del total del genoma, e incluso en regiones que se suelen considerar como «codificantes» algunos científicos creen que todavía hay que decodificar hasta un 10 por ciento, incluidas regiones que influyen en el envejecimiento.
En el relativamente breve período que ha transcurrido desde 2003, hemos descubierto que, dentro de la famosa doble hélice, había secuencias que no solo no estaban mapeadas, sino que además eran esenciales para nuestra vida. De hecho, muchos miles de secuencias han pasado desapercibidas porque los algoritmos originales para detectar genes se escribieron para desechar cualquier gen con menos de trescientos pares de bases. En realidad, los genes pueden tener hasta un mínimo de veintiún pares de bases y en la actualidad estamos descubriendo cientos por todo el genoma.
Estos genes les dicen a nuestras células que creen proteínas específicas, las cuales son los ladrillos de los procesos y los rasgos que constituyen la biología humana y las experiencias vitales. Y a medida que nos aproximamos a la identificación de la secuencia completa de nuestro ADN, nos acercamos también a tener un «mapa» de los genes que controlan casi toda nuestra existencia.
Sin embargo, una vez que completemos el código, seguiremos sin poder encontrar algo.
No encontraremos el gen responsable del envejecimiento.
Hemos descubierto genes que influyen en los síntomas del proceso del envejecimiento. Hemos descubierto genes de longevidad que controlan las defensas del organismo contra el envejecimiento y que ofrecen una forma de ralentizar dicho proceso a través de intervenciones naturales, farmacéuticas y tecnológicas. Pero, a diferencia de los oncogenes descubiertos en los años setenta del siglo pasado, que nos han proporcionado un buen objetivo para luchar contra el cáncer, no hemos identificado ni un solo gen que cause el envejecimiento. Ni lo identificaremos.
Porque nuestros genes no evolucionaron para causar el envejecimiento.
LA LEVADURA DEL EDÉN
Mi camino hasta formular la teoría del envejecimiento por pérdida de información fue muy largo. Y en gran medida se puede trazar hasta el trabajo de un científico que pasó sin pena ni gloria, pero que ayudó a fundamentar gran parte de la investigación en longevidad que se lleva a cabo hoy en día en todo el mundo.
Se llamaba Robert Mortimer y si algo se dijo de él con frecuencia después de su muerte fue que era «amable».
«Visionario» es otro adjetivo que le dedicaron. «Brillante», «inquisitivo» y «trabajador» también. Pero el ejemplo que les dio a sus colegas científicos lleva mucho tiempo inspirándome. Mortimer, que murió en 2007, tuvo un papel importantísimo a la hora de situar a la Saccharomyces cerevisiae, una levadura unicelular de aspecto humilde y con querencia por el dulce (su nombre significa «amante del azúcar»), en el lugar que se merecía como uno de los organismos de investigación más importantes del mundo.
Mortimer reunió miles de cepas de levaduras mutantes en su laboratorio, algunas de las cuales se habían desarrollado allí mismo, en la Universidad de California, en Berkeley. Podría haber financiado su investigación, y le habría sobrado, con el dinero cobrado a los miles de científicos a los que surtía a través de una especie de banco genético, el Yeast Genetic Stock Center. Sin embargo, cualquier persona, desde el estudiante más humilde hasta los profesores titulares de las instituciones de investigación mejor financiadas del mundo, tenía acceso al catálogo del centro y podía pedir cualquier cepa, que se le mandaba sin demora a cambio de los gastos de envío.
Y gracias a que lo convirtió en algo tan sencillo y barato, la investigación con levaduras floreció.
Cuando Mortimer empezó a trabajar con la S. cerevisiae junto con su colega biólogo John Johnston en la década de 1950, casi nadie tenía interés en la levadura. La mayoría creía que no podríamos aprender mucho de nuestros complejos seres estudiando hongos diminutos. Fue una ardua tarea convencer a la comunidad científica de que la levadura podía ser útil para algo más que hacer pan, elaborar cerveza o envejecer vino.
Lo que Mortimer y Johnston averiguaron, y lo que muchos otros empezaron a entender en los años posteriores, era que esas diminutas células de levadura no son tan distintas de nosotros. Pese a su tamaño, su composición genética y bioquímica es increíblemente compleja, lo que las convierte en un modelo excepcional para entender los procesos biológicos que sustentan la vida y que controlan la longevidad en organismos complejos como el nuestro. Si no terminas de creerte que una célula de levadura pueda decirnos algo acerca del cáncer, del alzhéimer, de las enfermedades raras o del envejecimiento, ten en cuenta que hay cinco premios Nobel de Fisiología o Medicina que recibieron el galardón por estudios genéticos con levadura, incluido el premio de 2009 por descubrir cómo las células contrarrestan el acortamiento de los telómeros, una de las marcas distintivas del envejecimiento.
El trabajo de Mortimer y Johnston, en particular un artículo trascendental en 1959 que demostraba que la longevidad de las células de levadura madres e hijas puede ser muy distinta, prepararía el camino para un cambio absolutamente revolucionario en la forma en la que percibimos los límites de la vida. Y para cuando Mortimer murió en 2007, había en el mundo alrededor de diez mil investigadores estudiando la levadura.
Sí, los humanos estamos separados de la levadura por mil millones de años de evolución, pero seguimos teniendo muchas cosas en común. S. cerevisiae comparte alrededor del 70 por ciento de nuestros genes. Y lo que hace con dichos genes no es tan distinto de lo que hacemos nosotros. Al igual que muchos humanos, las células de levadura se pasan la vida intentando hacer dos cosas: o comer o reproducirse. O tienen hambre o están excitadas. A medida que envejecen, de forma muy parecida a los humanos, se ralentizan y se vuelven más grandes, más orondas y menos fértiles. Pero, si bien los humanos realizan este proceso a lo largo de muchas décadas, las células de levadura lo experimentan en una semana. Eso las convierte en un punto de partida estupendo para emprender la labor de entender el envejecimiento.
De hecho, el potencial de que una humilde célula de levadura nos diga tanto acerca de nosotros, y que lo haga con relativa rapidez comparada con otros organismos de investigación, fue uno de los principales motivos por los que decidí empezar mi carrera profesional estudiando la S. cerevisiae. Además, huelen a pan recién hecho.
Conocí a Mortimer en Viena, en 1992, cuando yo tenía veintipocos años y asistí a la International Yeast Conference (sí, hay una convención internacional de la levadura) con los dos supervisores de mi doctorado: Ian Dawes, profesor australiano de la Universidad de Nueva Gales del Sur con aversión a las normas, y el profesor Richard Dickinson, un británico de la Universidad de Cardiff, en Gales, que las respetaba todas.
Mortimer estaba en Viena para hablar de un esfuerzo científico crucial: secuenciar el genoma de la levadura. Yo estaba allí para inspirarme. Y lo conseguí. Cualquier duda sobre mi decisión de dedicar mis primeros años como científico a un hongo unicelular se disipó en cuanto me encontré cara a cara con las personas que estaban generando una gran cantidad de conocimiento en un campo que apenas existía unas décadas atrás.
Fue poco después de esa conferencia cuando uno de los científicos más relevantes en el estudio de la levadura, Leonard Guarente, del MIT, vino a Sídney de vacaciones para visitar a Ian Dawes. Guarente y yo acabamos en la misma cena y me aseguré de sentarme enfrente de él.
Por aquel entonces yo era estudiante de doctorado y usaba la levadura para comprender un trastorno hereditario llamado «enfermedad de la orina con olor a jarabe de arce». Como podrás imaginar por el nombre, no es una enfermedad de la que la mayoría de la gente educada hablaría durante la cena. Sin embargo, Guarente entabló conmigo una discusión científica llena de curiosidad y un entusiasmo encantadores. La conversación derivó en su último proyecto (llevaba unos cuantos meses estudiando el envejecimiento de la levadura), un trabajo que se enraizaba en el mapa genético practicable que Mortimer había completado a mediados de los años setenta del siglo pasado.
Y con eso bastó. Me apasionaba comprender el envejecimiento, y lo de manejar células de levadura con un microscopio y un micromanipulador tenía su encanto. Eran habilidades esenciales para saber qué hacer con la levadura. Aquella noche, Guarente y yo estuvimos de acuerdo en algo: si no podíamos solucionar el problema del envejecimiento en la levadura, no teníamos la menor oportunidad de hacerlo en los seres humanos.
No solo quería trabajar con él: tenía que hacerlo.
Dawes le escribió para decirle que me interesaba muchísimo unirme a su laboratorio y que yo era «ducho en la mesa de trabajo».
«Será un placer trabajar con David», contestó él unas semanas después, de la misma manera que seguramente les contestaba a tantos aspirantes entusiastas. «Pero se lo tiene que financiar él», añadió. Más adelante descubrí que su emoción se debió a que me confundió con el otro estudiante que conoció durante la cena.
Tenía un pie dentro, pero contaba con pocas posibilidades. En aquella época, los extranjeros no se tenían en cuenta para las becas de posdoctorado más prestigiosas de Estados Unidos, pero yo insistí en hacer la entrevista y pagué de mi bolsillo el billete de avión a Boston. Me entrevistó un peso pesado en el campo de las células madre, Douglas Melton, para una beca de la Fundación Helen Hay Whitney, que ofrece apoyo económico a los estudiantes de posdoctorado en biomedicina desde 1947. Después de esperar mi turno a las puertas de su despacho junto con otros cuatro candidatos, llegó mi oportunidad. Era mi momento. No recuerdo estar nervioso. Supuse que, seguramente, no iba a conseguir la beca, de modo que fui a por todas.
Le hablé a Melton de mi ansia vital por entender el envejecimiento y por descubrir los «genes que dan la vida» antes de dibujar en su pizarra blanca cómo funcionan los genes y lo que haría durante los tres años siguientes si conseguía el dinero. Para demostrarle mi gratitud, le regalé una botella de vino tinto que le traje de Australia.
Después tuve claras dos cosas. La primera es que nunca lleves vino a una entrevista porque se puede considerar un intento de soborno. Y la segunda, que a Melton debió de gustarle lo que dije y cómo lo dije, porque volví a casa, conseguí la beca y luego me subí a otro avión de vuelta a Boston. Fue, sin duda alguna, el encuentro que más ha marcado mi vida.
Cuando llegué, en 1995, esperaba aumentar nuestra comprensión del envejecimiento estudiando el síndrome de Werner, una terrible enfermedad que aparece en menos de uno de cada cien mil nacimientos, con síntomas como la pérdida de masa corporal, arrugas, canas, pérdida de pelo, cataratas, osteoporosis, problemas coronarios y muchos otros indicios inconfundibles del envejecimiento. Y no me refiero en personas de setenta u ochenta años, sino en personas de treinta o cuarenta. La esperanza de vida de alguien con el síndrome de Werner es de cuarenta y seis años.
Sin embargo, dos semanas después de mi llegada a Estados Unidos, un equipo de investigación de la Universidad de Washington, encabezado por el sabio y alentador abuelo de la investigación en envejecimiento, George Martin, anunció que había encontrado el gen que, mutado, provoca el síndrome de Werner. En aquel momento, fue descorazonador que me hubieran «quitado la exclusiva», pero el descubrimiento me permitió dar un primer paso mucho mayor hacia mi verdadero objetivo. De hecho, se convirtió en un punto clave para formular la teoría del envejecimiento por pérdida de información.
Una vez identificado en seres humanos el gen de Werner, conocido como WRN, el siguiente paso era comprobar si el gen parecido de la levadura tenía la misma función. Si era así, podríamos usar la levadura para determinar con más rapidez la causa del síndrome de Werner y tal vez ayudarnos a entender mejor el envejecimiento en general. Entré en el despacho de Guarente para decirle que iba a empezar a estudiar el síndrome de Werner en la levadura y que así solucionaríamos el problema del envejecimiento.
En la levadura, el equivalente del gen WRN es el supresor de crecimiento lento 1 o SGS1, por sus siglas en inglés. Ya se sospechaba que el gen codificaba un tipo de enzima llamada «helicasa de ADN», que desenrolla las cadenas enrolladas de ADN antes de que se rompan. Las helicasas son especialmente importantes en las secuencias repetitivas de ADN que están predispuestas a enrollarse y a romperse. La funcionalidad de las proteínas, como las codificadas por el gen Werner, es vital, dado que la mitad de nuestro genoma es, de hecho, repetitivo.
A través de un proceso de intercambio de genes en el que se engaña a las células para que atrapen trozos de ADN adicionales, extrajimos el gen SGS1 funcional y lo cambiamos por una versión mutada. De hecho, estábamos intentando comprobar si era posible provocarle el síndrome de Werner a la levadura.
Después del cambio, la esperanza de vida de las células de levadura se redujo a la mitad. En circunstancias normales, esto no habría sido noticia. Muchos sucesos que nada tienen que ver con el envejecimiento, como que se la coma un ácaro, que se seque en una uva o que acabe en un horno, pueden acortar, y de hecho lo hacen, la vida de las células de levadura. Pero habíamos alterado su ADN, lo que podría haber cortocircuitado las células de mil formas distintas para provocarle una muerte temprana.
Sin embargo, las células no solo se morían: lo hacían después de un pronunciado deterioro de salud y de funcionamiento. A medida que los SGS1 mutantes envejecían, su ciclo celular se ralentizaba, se volvían más grandes. Genes tanto masculinos como femeninos de tipo «sexual» (descendientes del gen A) se activaron al mismo tiempo, de modo que eran estériles y no se pudieron reproducir. Esas eran las características principales del envejecimiento en la levadura. Y sucedía más deprisa en los mutantes que habíamos creado. Desde luego que parecía una versión del síndrome de Werner en la levadura.
Usando cadenas especializadas, coloreamos el ADN de azul y usamos rojo para el nucléolo, que se encuentra en el núcleo de todas las células eucariotas. Eso facilitó ver en el microscopio lo que sucedía a nivel celular.
Y era fascinante.
El nucléolo es una parte del núcleo en la que se encuentra el ADN ribosómico, o ADNr. Este se copia en el ARN ribosómico, el cual usan las enzimas ribosómicas para unir los aminoácidos que componen cada nueva proteína.
En las células SGS1 envejecidas, el nucléolo parecía haber explotado. En vez de una sola figura con forma curvada teñida de rojo y nadando en un mar azul, el nucléolo se había diseminado en media docena de pequeñas islas. Era trágico y hermoso. La imagen, que aparecería en el número de agosto de 1997 de la prestigiosa revista Science, sigue colgada en mi despacho.
Lo que sucedió a continuación fue fascinante y revelador al mismo tiempo. En respuesta al daño, como las ratas que respondían a la llamada del flautista de Hamelín, la proteína Sir2 (la primera sirtuina conocida, que está codificada por el gen SIR2 y desciende del gen B) se había alejado de los genes de tipo sexual que controlan la fertilidad para trasladarse al nucléolo.
Para mí era una imagen maravillosa, pero para la levadura era un problema. Sir2 tiene un trabajo importante: es un factor epigenético, una enzima que se coloca sobre los genes, envuelve el ADN y lo mantiene silenciado. A nivel molecular, la Sir2 lo consigue a través de su actividad enzimática, asegurándose de que los químicos denominados «acetilos» no se acumulen en las histonas y descompriman el ADN.
Cuando las sirtuinas dejaron los genes de tipo sexual (los que descienden del gen A que controlaba la fertilidad y la reproducción), las células mutantes se convirtieron en genes tanto masculinos como femeninos, haciendo que perdieran su identidad sexual de la misma manera que en las células viejas normales, pero mucho antes.
Al principio no comprendía por qué el nucléolo estaba explotando y mucho menos por qué las sirtuinas se movían hacia él a medida que las células envejecían. Me estuve comiendo la cabeza durante semanas con esta cuestión.
Y entonces, una noche, después de un largo día de trabajo en el laboratorio, me desperté con una idea.
Se me ocurrió en ese momento, entre el delirio provocado por la vigilia y el sueño profundo. El susurro de una idea. Un batiburrillo de palabras. Una imagen borrosa. Aunque eso bastó para que me despertase de pronto y me levantara.
Cogí el cuaderno y fui a la cocina. Allí, encorvado sobre la mesa a primera hora del 28 de octubre de 1996, empecé a escribir.
Una teoría sobre la senescencia replicativa en la levadura y otros organismos.
Estuve escribiendo alrededor de una hora, anotando ideas, garabateando, dibujando gráficos, formulando nuevas ecuaciones… Las observaciones científicas que antes no tenían sentido para mí empezaban a encajar en una imagen más amplia. El ADN roto provoca inestabilidad en el genoma, escribí, lo que distrae a la proteína Sir2, que a su vez cambia el epigenoma, haciendo que las células pierdan su identidad y se vuelvan estériles mientras intentaban reparar el daño. Eran los arañazos analógicos en los DVD digitales. Los cambios epigenéticos provocan el envejecimiento.
Había, me imaginé, un proceso singular que controlaba todos los demás. No un incontable número de cambios celulares individuales o de trastornos. Ni siquiera una serie de características que pudieran acatarse con una medicina apagafuegos. Había algo mayor, y más excepcional, que todo eso.
Eran los cimientos para entender el circuito de supervivencia y su papel en el envejecimiento.
Al día siguiente, le enseñé mis notas a Guarente. Estaba emocionado, tenía la sensación de que era la mejor idea que se me había ocurrido. Pero también estaba nervioso. Me daba miedo que Guarente encontrase una laguna en mi teoría y la echara por tierra. En cambio, repasó en silencio mi cuaderno, me hizo unas cuantas preguntas y me despachó con seis palabras.
—Me gusta. Ahora tienes que demostrarlo.
EL RECITAL
Para comprender la teoría del envejecimiento por pérdida de información, tenemos que volver al epigenoma, la parte de la célula que ayudan a controlar las sirtuinas.
De cerca, el epigenoma es más complejo y maravilloso que cualquier cosa que haya inventado el ser humano. Consiste en cadenas de ADN enrolladas alrededor de unas proteínas que actúan como carretes llamadas «histonas», que a su vez están integradas en circuitos mayores llamados «cromatinas», que a su vez están integrados en circuitos todavía mayores llamados «cromosomas».
Las sirtuinas les ordenan a las histonas que aten en corto el ADN, mientras que dejan que otras regiones den vueltas. De esta forma, algunos genes permanecen silenciados, mientras que a otros se accede a través de factores de transcripción que unen el ADN y que activan los genes. Se dice que los genes accesibles están en la eucromatina, mientras que los genes silenciados están en la heterocromatina. Al eliminar las etiquetas químicas en las histonas, las sirtuinas ayudan a evitar que los factores de transcripción se unan a los genes, convirtiendo la eucromatina en la heterocromatina.
Por supuesto, cada una de nuestras células tiene el mismo ADN, así que lo que diferencia una célula nerviosa de una epitelial es el epigenoma, el término colectivo para el sistema de control y las estructuras celulares que le dice a la célula qué genes debe activar y cuáles deben permanecer inactivos. Y esto, mucho más que los genes, es lo que controla casi toda nuestra vida.
Una de las mejores formas de visualizarlo es imaginar el genoma humano como un piano enorme. Cada gen es una tecla y cada tecla produce una nota. Y de un instrumento a otro, dependiendo del fabricante, de los materiales y de las circunstancias de la fabricación, el sonido varía, aunque se toquen de la misma manera. Estos son nuestros genes. Tenemos unos veinte mil, unos cuantos miles arriba o abajo.
Cada tecla también se puede tocar pianísimo o forte. Las notas también pueden ser sostenidas o allegretto. Para los virtuosos del piano, hay cientos de maneras de tocar cada tecla e infinidad de formas de tocarlas juntas, en acordes y combinaciones que crean música: jazz, ragtime, rock, reggae, vals, etc.
El pianista que lo hace posible es el epigenoma. A través del proceso de revelar nuestro ADN o de compactarlo con fuerza en paquetes de proteínas, y al marcar nuestros genes con etiquetas químicas denominadas «metilos» y «acetilos» compuestos de carbono, oxígeno e hidrógeno, el epigenoma usa nuestro genoma para crear la música de nuestra vida.
Sí, a veces el tamaño, la forma y la condición del piano dictaminan lo que el pianista puede lograr. Es difícil tocar un concierto con un piano de juguete de dieciocho teclas, y es infinitamente más difícil crear música bonita con un instrumento que lleva cincuenta años sin afinarse. De la misma manera, el genoma dictamina sin lugar a duda lo que puede hacer el epigenoma. Una oruga no puede convertirse en un ser humano, pero puede convertirse en mariposa gracias a los cambios en la expresión epigenética que suceden durante la metamorfosis, aunque su genoma nunca cambia. De igual modo, un hijo de dos padres de un largo linaje de personas de pelo negro y ojos castaños no tiene muchas probabilidades de tener el pelo rubio y los ojos azules, pero unos ratones agutí gemelos en el laboratorio pueden acabar teniendo el pelaje marrón o dorado, dependiendo de la cantidad de gen agutí que se activa durante la gestación gracias a la influencia ambiental en el epigenoma, como el ácido fólico, la vitamina B12, la genisteína de la soja o la toxina bisfenol A.
De manera similar, entre los gemelos humanos monocigóticos, las fuerzas epigenéticas pueden llevar a dos personas con el mismo genoma en direcciones totalmente opuestas. Incluso pueden hacer que envejezcan de forma distinta. Puedes verlo con claridad si miras dos fotos juntas de unos gemelos, de los que uno es fumador y el otro no; su ADN sigue siendo en gran parte igual, pero los fumadores tienen más ojeras, más papada y más arrugas alrededor de los ojos y de la boca. No son mayores, pero es evidente que han envejecido más deprisa. Los estudios en gemelos idénticos estiman que la influencia genética en la longevidad está entre un 10 y un 25 por ciento, algo que, se mire como se mire, es sorprendentemente bajo.
Nuestro ADN no es nuestro destino.
Ahora imagina que estás en una sala de conciertos. Una virtuosa del piano está sentada a un reluciente Steinway de cola. El concierto empieza. La música es preciosa, arrebatadora. Todo es perfecto.
Pero entonces, a los pocos minutos de empezar, la pianista toca mal una tecla. La primera vez que sucede es casi imperceptible, tal vez un re en un acorde que no lo necesita. Embutido en tantas notas tocadas a la perfección, oculto en un acorde impecable de una melodía perfecta, no es motivo de preocupación. Pero luego, a los pocos minutos, sucede otra vez. Y luego la frecuencia cada vez es mayor, una y otra vez.
Es importante recordar que al piano no le pasa nada y que la pianista está tocando la mayoría de las notas establecidas por el compositor. El asunto es que también está tocando algunas notas de más. Al principio es solo un poco molesto, pero con el transcurso del tiempo empieza a ser inquietante. Al final, arruina el concierto. De hecho, uno asumiría que le pasa algo a la pianista. Tal vez incluso alguien suba al escenario para asegurarse de que se encuentra bien.
El ruido epigenético provoca el mismo caos. Se compone en gran parte de ataques muy perturbadores a las células, como el ADN roto, tal y como sucedió en el circuito de supervivencia original de M. superstes y en las viejas células de levadura que perdieron la fertilidad. Y esto, según la teoría del envejecimiento por pérdida de información, es el motivo de que envejezcamos. Por eso nos salen canas. Por eso se nos arruga la piel. Por eso empiezan a dolernos las articulaciones. Es más, por eso suceden todas y cada una de las marcas distintivas del envejecimiento, desde el agotamiento de las célula madre y la senescencia celular hasta la disfunción mitocondrial y el acortamiento rápido de los telómeros.
Reconozco que es una teoría audaz. Y la fortaleza de una teoría se basa en lo bien que predice los resultados de experimentos rigurosos, a menudo millones; en el número de fenómenos que puede explicar; y en su sencillez. La teoría era sencilla y explicaba mucho. Como buenos científicos, solo nos quedaba esforzarnos para desacreditarla y ver cuánto tiempo sobrevivía.
Para empezar, Guarente y yo teníamos que echarle un vistazo al ADN de algunas levaduras.
Usamos una técnica llamada Southern blot, un método para separar el ADN basado en su tamaño y su configuración, tras lo cual se resalta con una sonda radiactiva de ADN. En el primer experimento nos dimos cuenta de algo espectacular. Normalmente, el ADNr de una célula de levadura que se hace visible gracias a esa técnica está bien compactado, como una cuerda nueva enrollada, con unas cuantas espirales de ADN muy compactado. Sin embargo, el ADNr de las células de levadura que creamos en el laboratorio (las mutantes de Werner que parecían envejecer muy deprisa) se estaba descomprimiendo a toda velocidad, como una bolsa llena de lana sellada al vacío que se hubiera roto de repente.
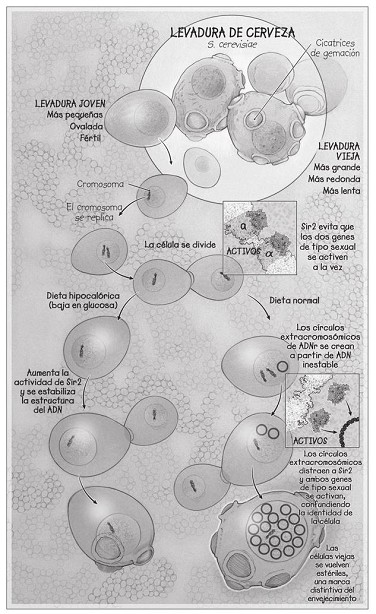
El ADNr se hallaba sumido en el caos. Parecía que el genoma se estaba fragmentando. El ADN se estaba recombinando y amplificando y aparecía en la prueba de Southern blot como puntos negros y espirales ralas, dependiendo de lo compactadas y comprimidas que estuvieran. Llamamos a esas espirales «círculos extracromosómicos ribosómicos de ADN» o ERC (por sus siglas en inglés), que se iban acumulando a medida que las mutantes envejecían.
Si de verdad hubiéramos inducido el envejecimiento, deberíamos de haber visto el mismo patrón en las células de levadura que habían envejecido de forma normal.
La edad de una célula de levadura no se puede saber por las velas de cumpleaños. Simplemente, no duran tanto. En cambio, el envejecimiento de la levadura se mide contando el número de veces que una célula madre se divide para producir células hija. En la mayoría de los casos, una célula de levadura se divide unas veinticinco veces antes de morir. Sin embargo, eso hace que conseguir células de levadura viejas sea particularmente difícil, porque, cuando una célula de levadura normal muere, está rodeada de 225 (33 millones) de descendientes.
Tardamos una semana de trabajo, muchas noches en vela y un montón de cafeína en recolectar suficientes células viejas normales. Al día siguiente, mientras revelaba la película para visualizar el ADNr, lo que vi me dejó de piedra.
Al igual que las mutantes, las células viejas de levadura normales estaban llenas de círculos extracromosómicos de ADNr.
¡Eureka! No era una prueba, porque un buen científico nunca tiene pruebas de nada, pero sí la primera confirmación sustancial de una teoría, los cimientos sobre los que construir nuevos descubrimientos en los años venideros.
La primera predicción comprobable era que, si poníamos un ERC en células de levadura muy jóvenes (y diseñamos un truco genético para lograrlo), los ERC se multiplicarían y distraerían a las sirtuinas, y las células de levadura envejecerían de forma prematura, se volverían estériles y morirían jóvenes… Y así fue. Publicamos aquel trabajo en diciembre de 1997, en la revista científica Cell, y la noticia dio la vuelta al mundo: «Unos científicos descubren la causa del envejecimiento».
Fue justo entonces cuando Matt Kaeberlein, un estudiante de doctorado en aquella época, llegó al laboratorio. Su primer experimento fue introducir una copia extra de SIR2 en el genoma de células de levadura para comprobar si estabilizaría el genoma de la levadura y retrasaría el envejecimiento. Cuando añadió SIR2, se evitaron los ERC y comprobó que la esperanza de vida de las células de levadura aumentó en un 30 por ciento, tal como habíamos esperado. Nuestra hipótesis parecía sobrevivir al escrutinio: la causa fundamental y problemática de la esterilidad y del envejecimiento en la levadura era la inestabilidad inherente del genoma.
Lo que surgió de aquellos primeros resultados con levadura, y de otra década de investigación y de pruebas en células de mamíferos, fue una forma revolucionaria de entender el envejecimiento, una teoría de la pérdida de información que reconciliaría los factores del envejecimiento que parecían tan dispares en un modelo universal de vida y muerte. Es algo semejante a lo siguiente:
Juventud ➝ ADN roto ➝ Inestabilidad del genoma ➝ Alteración en la compactación del ADN y de la regulación de los genes (el epigenoma) ➝ Pérdida de identidad celular ➝ Senescencia celular ➝ Enfermedad ➝ Muerte.
Las connotaciones eran profundas: si pudiéramos intervenir en algunos de estos pasos, podríamos ayudar a las personas a vivir más tiempo.
Pero ¿y si pudiéramos intervenir en todas? ¿Podríamos detener el envejecimiento?
Las teorías hay que probarlas, volver a probarlas y probarlas otra vez, y no por un solo científico, sino por muchos. Y en ese aspecto, tuve la suerte de formar parte de un equipo de investigación que incluía a algunos de los científicos más brillantes y perspicaces del mundo.
Estaba Lenny Guarente, nuestro infatigable mentor. También estaba Brian Kennedy, que empezó el proyecto sobre el envejecimiento de la levadura en el laboratorio y que ha tenido un papel importantísimo a la hora de entender las enfermedades de envejecimiento prematuro y el impacto de los genes y de las moléculas que aumentan la salud y la longevidad en los organismos modelo. También estaban Monica Gotta y Susan Gasser, de la Universidad de Ginebra, que ahora son dos de las investigadoras más relevantes en el campo de la regulación genética; Shin-ichiro Imai, ahora profesor en la Universidad de Washington, que descubrió que las sirtuinas son enzimas que utilizan NAD y ahora está investigando cómo controla el cuerpo las sirtuinas; Kevin Mills, que dirigía un laboratorio en Maine y que después se convirtió en cofundador e investigador jefe en Cyteir Therapeutics, que desarrolla novedosas formas de luchar contra el cáncer y las enfermedades autoinmunes; Nicanor Austriaco, que empezó el proyecto con Brian y que ahora enseña biología y teología en Providence College, una gran combinación; Tod Smeal, investigador jefe de biología del cáncer en la multinacional farmacéutica Eli Lilly; David Lombard, que ahora es investigador en el campo del envejecimiento en la Universidad de Míchigan; Matt Kaeberlein, profesor en la Universidad de Washington, que está probando moléculas en la longevidad de los perros; David McNabb, cuyo laboratorio en la Universidad de Arkansas ha hecho descubrimientos clave que salvan vidas sobre los patógenos micóticos; Bradley Johnson, experto en envejecimiento humano y cáncer en la Universidad de Pensilvania; y Mala Murthy, una prominente neurocientífica que ahora está en Princeton.
Una y otra vez he disfrutado del enorme privilegio de trabajar con las personas con las que he trabajado. Y sobre todo en el laboratorio de Guarente en el MIT. Era el equipo perfecto y a menudo me sentía honrado por las personas que me rodeaban.
Cuando empecé mi carrera en este campo, soñaba con publicar al menos un estudio en una revista prestigiosa. A lo largo de aquellos años, nuestro grupo publicaba uno cada pocos meses.
Demostramos que la redistribución de Sir2 hacia el nucléolo es una respuesta a numerosas fracturas en el ADN, que suceden cuando los ERC se multiplican y se reintegran en el genoma, o cuando se juntan entre sí para formar un ERC enorme. Cuando Sir2 se desplaza para combatir la inestabilidad del ADN, vuelve estériles a las células de levadura hinchadas y viejas. Ese fue el primer paso del circuito de supervivencia, aunque en aquel momento no teníamos ni idea de que era tan antiguo y tan esencial para nuestra existencia como demostró ser.
Le contamos al mundo que podíamos provocarle a la levadura un síndrome parecido al de Werner, dando lugar a nucléolos que explotaban. Describimos las formas en las que los mutantes de SGS1, los que habíamos saturado con el equivalente en la levadura de la mutación del síndrome de Werner, acumulaban ERC más deprisa, lo que llevaba a un envejecimiento prematuro y a una vida más corta. Al demostrar que, si añades un ERC a células jóvenes, estas envejecen de forma prematura, obtuvimos la prueba crucial de que los ERC no solo suceden durante el envejecimiento, sino también de que lo causan. Y al romper de forma artificial el ADN en la célula y observar la respuesta celular, demostramos por qué se mueven las sirtuinas: para ayudar a reparar el ADN. Eso demostró ser el segundo paso del circuito de supervivencia. El daño en el ADN que propició el aumento de los ERC hacía que Sir2 se alejara de los genes de tipo sexual, que se volvían estériles, una marca del envejecimiento de la levadura.
Era ruido epigenético en su máxima expresión.
Llevó otros veinte años comprobar si esos descubrimientos sobre la levadura eran relevantes para organismos más complejos. Los mamíferos tenemos siete genes de sirtuina que han desarrollado una variedad de funciones más allá de lo que el simple SIR2 puede hacer. Tres de ellos, SIRT1, SIRT6 y SIRT7, son críticos para el control del epigenoma y la reparación del ADN. Los otros, SIRT3, SIRT4 y SIRT5, residen en la mitocondria, donde controlan el metabolismo de la energía, mientras que SIRT2 da vueltas alrededor del citoplasma, donde controla la división celular y la producción de óvulos sanos.
Encontramos muchas pistas por el camino. Stephen Helfand, de la Universidad de Brown, demostró que añadir copias extra del gen dSir2 a la mosca de la fruta suprime el ruido epigenético y aumenta su longevidad. Descubrimos que, en los mamíferos, SIRT1 se aleja de los genes silenciados para ayudar a reparar el ADN roto en células de ratones y de seres humanos. Pero no supimos con certeza hasta qué punto se conservaba el circuito de supervivencia entre las levaduras y los seres humanos hasta 2017, cuando el equipo de Eva Bober, del Instituto Max Planck para la Investigación Cardíaca y Pulmonar en Bad Nauheim, en Alemania, anunció que las sirtuinas estabilizan el ADNr humano. Después, en 2018, Katrin Chua, de la Universidad de Stanford, descubrió que, al estabilizar el ADNr humano, las sirtuinas evitan la senescencia, que es básicamente la misma función antienvejecimiento de las sirtuinas en la levadura que habíamos descubierto veinte años antes.
Fue una revelación sorprendente: más de mil millones de años nos separan de la levadura, pero, en esencia, el circuito no había cambiado.
Sin embargo, cuando por fin salieron a la luz esos descubrimientos, yo ya tenía claro que el ruido epigenético seguramente fuera un catalizador del envejecimiento humano. Dos décadas de investigación ya nos encaminaban en esa dirección.
En 1999, crucé el río para dejar el MIT y trasladarme a la Escuela de Medicina de Harvard, donde organicé otro laboratorio para estudiar el envejecimiento. Allí esperaba dar respuesta a una nueva pregunta que me llevaba rondando la cabeza un tiempo.
Me había dado cuenta de que las células de levadura alimentadas con menos cantidad de azúcar no solo vivían más tiempo, sino que además su ADNr era excepcionalmente compacto, lo que retrasaba de forma significativa la inevitable acumulación de ERC, las catastróficas roturas de ADN, la explosión del nucléolo, la esterilidad y la muerte.
¿Por qué sucedía esto?
EL CIRCUITO DE SUPERVIVENCIA SE HACE MAYOR
Nuestro ADN soporta asaltos constantes. De media, cada uno de nuestros cuarenta y seis cromosomas se rompe de alguna manera cada vez que una célula copia su ADN, lo que supone más de dos billones de roturas al día en nuestro cuerpo. Y eso solo contando las roturas que se producen durante la replicación. Otras las provocan la radiación natural, los químicos de nuestro entorno, y los rayos X y las tomografías que nos hacen.
Si no pudiéramos reparar nuestro ADN, no duraríamos demasiado. Por ese motivo, en el primordio, los ancestros de todos los seres vivos de este planeta evolucionaron para percibir el daño en el ADN, para ralentizar el crecimiento celular y para dedicar energía a la reparación del ADN hasta que estuviera arreglado. Es lo que denomino «circuito de supervivencia».
Desde el trabajo con la levadura, han ido apareciendo nuevas pruebas de que las levaduras no son tan distintas de nosotros. En 2003, Michael McBurney, de la Universidad de Ottawa, en Canadá, descubrió que los embriones de ratas manipulados para ser incapaces de producir una de las siete enzimas de la sirtuina, SIRT1, no podían pasar del decimocuarto día de desarrollo, unos dos tercios del período de gestación de un ratón. Entre los motivos, según contó el equipo en la revista Cancer Cell, se encontraba la escasa capacidad de respuesta y reparación del daño en el ADN. En 2006, Frederick Alt, Katrin Chua y Raul Mostovslavsky, de Harvard, demostraron que los ratones creados para que no tuvieran SIRT6 presentaron los signos típicos del envejecimiento con más rapidez y vivieron menos. Cuando los científicos eliminaban la habilidad de una célula para crear esta proteína vital, la célula perdía su capacidad para reparar las roturas del ADN bicatenario, tal y como demostramos nosotros con la levadura en 1999.
Si eres escéptico, y deberías serlo, podrías suponer que estos ratones con SIRT mutante estaban enfermos y, por tanto, vivían menos. Pero añadir más copias de los genes de sirtuina SIRT1 y SIRT6 hace justo lo contrario: aumenta la salud y la vida de los ratones, de la misma manera que cuando se añaden copias extra a la levadura de su gen SIR2. Estos descubrimientos se los debemos a dos de mis anteriores colegas, Shin-ichiro Imai, mi antiguo compañero de copas en el laboratorio de Guarente, y Haim Cohen, mi primer investigador de posdoctorado en Harvard.
En la levadura, habíamos demostrado que las roturas de ADN provocan que las sirtuinas se alejen de los genes de tipo sexual silenciados, haciendo que las células viejas se vuelvan estériles. Era un sistema sencillo y lo desciframos en unos pocos años.
Pero ¿está provocando el circuito de supervivencia el envejecimiento en los mamíferos? ¿Qué partes del sistema han sobrevivido estos miles de millones de años y cuáles son específicas de la levadura? Esas preguntas están en el abismo del conocimiento humano ahora mismo, pero las respuestas empiezan a revelarse.
Lo que yo sugiero es que el gen SIR2, en el caso de la levadura, y los genes SIRT, en los mamíferos, descienden todos del gen B, el silenciador de genes original en M. superstes. Su trabajo original era silenciar el gen que controlaba la reproducción.
En los mamíferos, las sirtuinas han desarrollado una serie de funciones nuevas y no solo actúan como controladoras de la fertilidad (aunque siguen siéndolo). Eliminan los acetilos de cientos de proteínas en las células: las histonas, sí, pero también proteínas que controlan la división celular, la supervivencia celular, la reparación del ADN, la inflamación, el metabolismo de la glucosa, la mitocondria y otras muchas funciones.
He llegado a considerar a las sirtuinas como las jefas de un polifacético cuerpo de respuesta ante desastres; envían a una variedad de equipos especializados de emergencias para encargarse de la estabilidad del ADN, su reparación, la supervivencia celular, el metabolismo y la comunicación entre las células. En cierto sentido, es como el centro de mando de los millares de trabajadores especializados que llegaron a Luisiana y a Mississippi después del paso del huracán Katrina en 2005. La mayoría de los trabajadores no era de la zona, pero fueron allí, hicieron lo que pudieron para arreglar el desastre y luego regresaron a casa. Antes de volver a su vida normal, algunos estuvieron trabajando en los barrios destrozados por el huracán unos días y otros, semanas. Y para la mayoría no fue la primera vez que hacían algo así ni sería la última. Cada vez que hay un desastre que afecta a los servicios básicos, se unen para ayudar.
Cuando están en casa, esta gente hace las cosas típicas que se hacen cuando uno está en casa: pagar las facturas, cortar el césped, entrenar a un equipo de béisbol o lo que sea. Pero cuando están fuera de casa, ayudando a evitar que lugares como la Costa del Golfo caigan en la anarquía (un estado que habría tenido consecuencias desastrosas para el resto del país), muchas de esas cosas se tienen que dejar en suspenso.
Cuando las sirtuinas abandonan sus prioridades normales para dedicarse a la reparación del ADN, su función epigenética en casa se detiene por un tiempo. Después, cuando el daño está reparado y vuelven a su hogar, retoman su tarea habitual: controlar los genes y asegurarse de que la célula mantiene su identidad y su función óptima.
Pero ¿qué sucede cuando hay una emergencia tras otra? ¿Un huracán tras otro? ¿Un terremoto tras otro? Las cuadrillas de reparadores están fuera de casa mucho tiempo y las cosas que suelen hacer se les acumulan. Las facturas se quedan sin pagar y generan intereses y luego comienzan a llegar los cobradores de deudas. El césped está demasiado alto y el presidente de la comunidad empieza a mandar enseguida mensajes agresivos. El equipo de béisbol se queda sin entrenador y se convierte en el colista de la liga. Y, sobre todo, una de las cosas más importantes que hacen en casa, reproducirse, no sucede. Esta forma de hormesis, el circuito de supervivencia original, está bien para mantener vivos a los organismos a corto plazo. Pero, a diferencia de las moléculas de la longevidad, que solo copian la hormesis al manipular sirtuinas, mTOR o AMPK, y envían tropas a falsas emergencias, las reales crean daños potencialmente mortales.
¿Qué podría provocar tantas emergencias? El daño en el ADN. ¿Y qué provoca eso? En fin, a lo largo del tiempo, la vida misma. Químicos malos. La radiación. Incluso la copia normal del ADN. Estas son las causas que creemos responsables del envejecimiento, pero hay que cambiar sutilmente, aunque es vital hacerlo, la forma en la que pensamos. No se trata tanto de que las sirtuinas están saturadas, aunque seguramente lo estén cuando te quemas por tomar el sol o te haces una radiografía; lo que sucede a diario es que las sirtuinas y los colaboradores que controlan el epigenoma no siempre encuentran el camino de vuelta a su lugar de origen después de que los llamen para una emergencia. Es como si unos cuantos trabajadores de los servicios de emergencia que acudieron a reparar los daños sufridos en la Costa del Golfo por el Katrina hubieran perdido la dirección de su casa. Luego el desastre se repite una y otra vez y tienen que salir de nuevo.
Cada vez que los factores epigenéticos dejan el genoma para encargarse de los daños, los genes que deberían estar inactivos se activan, y viceversa. Cada vez que se detienen en el genoma, hacen lo mismo, alterando así el epigenoma de formas nunca contempladas cuando fueron creados.
Las células pierden su identidad y empiezan a funcionar mal. Surge el caos, que se materializa como envejecimiento. Este es el ruido epigenético que se encuentra en el corazón de nuestra teoría unificada.
¿Cómo desactiva realmente el gen SIR2 los genes? SIR2 es el código de una proteína especializada llamada «histona deacetilasa», o HDAC, que a nivel enzimático elimina las etiquetas de acetilo de las histonas, que, tal como recordarás, hace que el ADN se compacte, evitando así que se transcriba en el ARN.
Cuando la enzima Sir2 está sobre los genes de tipo sexual, estos permanecen silenciados y la célula sigue emparejándose y reproduciéndose. Pero, cada vez que se produce una rotura en el ADN, se llama a Sir2 para que elimine las etiquetas de acetilo de las histonas en la rotura del ADN. Eso compacta las histonas para evitar que se recorte el ADN deshilachado y para ayudar a reclutar otras proteínas reparadoras. Una vez que la reparación del ADN está terminada, la mayor parte de la proteína Sir2 vuelve a los genes de tipo sexual para silenciarlos y restaurar la fertilidad. Por supuesto, lo hace si no hay otra emergencia, como sucede con la inestabilidad masiva del genoma que se produce cuando los ERC se acumulan en los nucléolos de las células viejas de levadura.
Para que el circuito de supervivencia funcione y provoque el envejecimiento, Sir2 y otros reguladores epigenéticos tienen que darse en «cantidades limitadas». Es decir, la célula no crea suficiente proteína Sir2 para silenciar los genes de tipo sexual a la vez que repara el ADN roto; tiene que trasladar Sir2 de un lugar a otro en función de las necesidades. Por eso, añadir una copia extra del gen SIR2 aumenta la esperanza de vida y retrasa la infertilidad: las células cuentan con suficiente Sir2 para reparar las roturas de ADN y también para silenciar los genes de tipo sexual.
A lo largo de los últimos miles de millones de años, es de suponer que millones de células de levadura han mutado de forma espontánea para crear más Sir2, pero murieron porque no tenían ventaja sobre otras células de levadura. Vivir para veintiocho divisiones no suponía ventaja alguna sobre las que vivían para veinticuatro; dado que Sir2 gasta energía, tener más de esta proteína incluso podría haber sido una desventaja. Sin embargo, en el laboratorio nosotros no nos percatamos de desventaja alguna, porque la levadura recibe más azúcar de la que puede ingerir. Al añadir copias extra del gen SIR2, les dimos a las células de levadura lo que la evolución no consiguió proporcionarles.
Si la teoría de la pérdida de información es correcta (que el envejecimiento está causado por señaladores epigenéticos saturados que responden a ataques y daños celulares), no importa tanto el lugar exacto donde sucede dicho daño. Lo que importa es que se está produciendo el daño y que las sirtuinas dan vueltas por todas partes para repararlo, dejando de lado sus responsabilidades habituales y a veces volviendo a otros puntos del genoma donde silencian genes que se supone que no tienen que silenciar. Es el equivalente celular de distraer al pianista celular.
Para demostrarlo, tuvimos que romper ADN de ratones.
No es difícil romper ADN a propósito. Se puede hacer con métodos de cizallamiento mecánico, así como con quimioterapia o con rayos X.
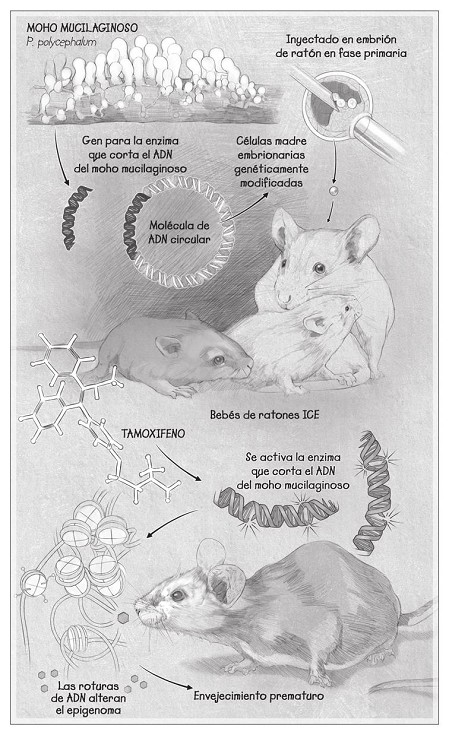
Pero teníamos que hacerlo con precisión, de un modo que no creara mutaciones ni que influyera en regiones que afectan a alguna función celular. En resumen, teníamos que atacar los páramos del genoma. Para hacerlo, echamos mano de un gen parecido a Cas9, la técnica de edición genética CRISPR de las bacterias que corta el ADN en puntos concretos.
La enzima escogida para nuestros experimentos procede de un moho amarillento y baboso llamado Physarum polycephalum, que literalmente significa «moho de muchas cabezas». La mayoría de los científicos cree que este gen, llamado I-PpoI, es un parásito que solo sirve para copiarse a sí mismo. Cuando corta el genoma del moho, se inserta otra copia de I-PpoI. Es el epítome de un gen egoísta.
Esto sucede en el moho mucilaginoso, su hábitat natural. Pero, cuando el I-PpoI se encuentra en una célula de ratón, no tiene toda la maquinaria del moho mucilaginoso para copiarse. De modo que da vueltas y corta el ADN solo en unos pocos puntos del genoma del ratón, sin proceso de copia. En cambio, la célula no tiene problemas a la hora de pegar las hebras de ADN otra vez, sin dejar mutaciones, que es justo lo que estábamos buscando para activar el circuito de supervivencia y distraer a las sirtuinas. Los genes de edición del ADN, como Cas9 e I-PpoI, son regalos de la naturaleza para la ciencia.
Para crear un ratón con el que poner a prueba la teoría de la pérdida de información, insertamos I-PpoI en una molécula de ADN circular llamada «plásmido», junto con todos los elementos reguladores del ADN necesario para controlar el gen, y luego insertamos ese ADN en el genoma de una línea de células madre embrionarias de un ratón que estábamos cultivando en placas de Petri en el laboratorio. Después inyectamos las células madre genéticamente modificadas en un embrión de ratón de noventa células que se llama «blastocito», lo implantamos en el útero de un ratón hembra y esperamos unos veinte días a que apareciera el bebé ratón.
Esto parece muy complicado, pero no lo es. Después de un poco de práctica, un estudiante universitario puede hacerlo. Es algo tan habitual hoy en día que puedes pedir un ratón específico por catálogo o incluso pagarle a una empresa para que te cree uno de acuerdo con tus especificaciones.
Los bebés ratones nacieron perfectamente normales, como era de esperar, dado que la enzima que corta estaba desactivada a esas alturas. Los llamamos de forma cariñosa «ratones ICE», siendo ICE «cambios inducibles en el epigenoma» (por sus siglas en inglés). Lo de «inducible» es vital, porque estos ratones no tienen nada de especial hasta que les damos una dosis baja de tamoxifeno. Se trata de un bloqueador de estrógenos que se suele emplear para tratar el cáncer en seres humanos, pero en este caso habíamos alterado el ratón de modo que el tamoxifeno activara el gen I-PpoI. La enzima se pondría a trabajar, cortando el genoma y sobrecargando ligeramente el circuito de supervivencia, sin matar célula alguna. Y, dado que el tamoxifeno tiene una vida de un par de días, eliminarlo de la comida de los ratones deshabilitaría los cortes en el genoma.
Los ratones quizá hubieran muerto. O desarrollado tumores. O podrían estar en perfecto estado, como si solo les hubiéramos hecho una radiografía dental. Nadie había hecho algo parecido en ratones, así que no lo sabíamos. Pero, si nuestra hipótesis acerca de la inestabilidad epigenética y del envejecimiento era correcta, el tamoxifeno actuaría como la poción que Fred y George Weasley se bebieron en Harry Potter y el cáliz de fuego.
Y funcionó. Fue como magia.
Durante el tratamiento, los ratones estuvieron bien, ajenos a los cortes de ADN y a la distracción de las sirtuinas. Pero, unos meses más tarde, recibí una llamada de una investigadora de posdoctorado que se ocupaba de nuestros animales de laboratorio mientras yo estaba en el de Australia.
—Uno de los ratones está muy enfermo —me dijo—. Creo que vamos a tener que sacrificarlo.
Le pedí que me enviara una foto del ratón del que me hablaba. Cuando me llegó al móvil, se me escapó la carcajada.
—No es un ratón enfermo —repliqué—. Es un ratón viejo.
—David —insistió ella—, creo que te equivocas. Aquí dice que es la hermana de los otros ratones de la jaula y los demás están perfectamente.
Su confusión era comprensible. Con dieciséis meses, un ratón de laboratorio normal todavía tiene el pelo fuerte, la cola activa, el cuerpo musculoso, las orejas erguidas y los ojos despejados. Un ratón ICE tratado con tamoxifeno tenía el pelo ralo y canoso, el cuerpo encogido, las orejas muy delgadas y los ojos velados.
Recuerda que no hicimos nada para cambiar el genoma. Solo habíamos roto el ADN de los ratones en puntos seleccionados donde no hay genes y obligamos a la célula a pegarlos, o «ligarlos», de nuevo. Para asegurarnos, más adelante también rompimos el ADN en otros puntos, con los mismos resultados. Esas roturas habían inducido una respuesta de las sirtuinas. Cuando estos trabajadores se pusieron manos a la obra, su ausencia en sus puestos habituales y su presencia en otras partes del genoma alteró las formas en las que muchos de los genes se expresaban en el momento inoportuno.
Esos hallazgos estaban en línea con los descubrimientos llevados a cabo por Trey Ideker y Kang Zhang, en la Universidad de California en San Diego, y por Steve Horvath, en la Universidad de California en Los Ángeles. El nombre de Steve caló y hoy su apellido se usa para denominar el «reloj de Horvath», una forma exacta de estimar la edad biológica de alguien gracias a la medición de miles de marcas epigenéticas en el ADN, llamada «metilación». Solemos pensar que el envejecimiento es algo que empieza a sucedernos en la mediana edad, porque es cuando comenzamos a ver cambios significativos en nuestro cuerpo. Pero el reloj de Horvath empieza a contar el tiempo desde que nacemos. Los ratones también tienen un reloj epigenético. ¿Eran mayores los ratones ICE que sus hermanos? Sí, lo eran… alrededor de un 50 por ciento mayores.
Habíamos encontrado lo que le daba cuerda al reloj maestro de la vida.
Si lo pensamos de otra manera, habíamos arañado el DVD de la vida un 50 por ciento más rápido de lo normal. El código digital que es, y era, el plano básico de nuestros ratones era el mismo de siempre, pero la máquina analógica construida para leer dicho código era capaz de captar solo retazos de los datos.
Aquí está lo más sobresaliente: podíamos envejecer ratones sin afectar a ninguna de las causas que más solían darse por sentado en el envejecimiento. No habíamos obligado a sus células a mutar, ni habíamos tocado sus telómeros, ni habíamos alterado sus mitocondrias ni habíamos agotado directamente sus células madre. Sin embargo, los ratones ICE perdían masa corporal, así como mitocondria y fuerza muscular, además de sufrir un aumento de cataratas, artritis, demencia, pérdida ósea y fragilidad.
Todos los síntomas del envejecimiento (y los trastornos que empujan a los ratones, al igual que a los seres humanos, hacia el precipicio de la muerte) los provocaban no ya la mutación, sino los cambios epigenéticos resultantes de las señales de daño en el ADN.
No les habíamos provocado a los ratones todas esas cosas: les habíamos provocado el envejecimiento.
Y si puedes provocar algo, lo puedes eliminar.
FRUTA DEL MISMO ÁRBOL
Como si fueran las retorcidas manos de unos zombis gigantes que se abren paso a través del suelo rocoso, los antiquísimos pinos longevos de las White Mountains, en California, crean figuras fantasmagóricas recortadas en el nebuloso sol del amanecer.
Los más antiguos llevan aquí desde antes de que se construyeran las pirámides de Egipto o Stonehenge y antes de que los mamuts lanudos se extinguieran. Han compartido este planeta con Moisés, Jesús, Mahoma y el primer Buda. A más de tres mil metros del nivel del mar, añadiendo milésimas de milímetros a sus retorcidos troncos cada año, desafiando las tormentas eléctricas y las recurrentes sequías, son el epítome de la perseverancia.
Qué fácil es quedarse anonadado frente a estos seres tan antiguos y enormes. Qué fácil es dejarse hechizar por su poder y su majestuosidad. Qué fácil es mirarlos embobado, sin más. Pero hay otra forma de ver a estos patriarcas antediluvianos, una más difícil, pero que todos los seres vivos de este planeta deberíamos aplicar: como los maestros que son para nosotros.
Al fin y al cabo, los pinos longevos son nuestros primos eucariotas. Casi la mitad de sus genes son parientes cercanos de los nuestros.
Pero no envejecen.
Por supuesto que suman años a su vida, miles y miles, marcados por anillos casi microscópicos que se esconden bajo su densa madera, aunque también se graban en su tamaño, su forma y su composición química eventos climatológicos del pasado, como cuando la erupción del Krakatoa lanzó una nube de ceniza que dio la vuelta al mundo en 1883, dejando un difuso anillo de crecimiento en 1884 y 1885, apenas a un centímetro del anillo de corteza exterior que marca nuestra época actual.
Sin embargo, pese al transcurso de tantos miles de años, sus células no parecen haber acusado deterioro alguno en cuanto a su función. Los científicos lo denominan «senescencia insignificante». De hecho, cuando un equipo del Instituto de Genética Forestal emprendió la búsqueda de signos de envejecimiento celular, estudiando pinos longevos de entre 23 y 4.713 años de antigüedad, no encontraron nada. Entre los árboles nuevos y los antiguos, su estudio de 2001 no descubrió diferencias significativas ni en los sistemas de transportación química, ni en la proporción del crecimiento de los brotes, ni en la calidad del polen que producían, ni en el tamaño de sus semillas ni en la forma en la que dichas semillas germinaban.
Los científicos también buscaron mutaciones deletéreas, esto es, la clase de mutaciones que muchos científicos de la época esperaban que fueran la causa principal del envejecimiento. No encontraron nada. Tengo la impresión de que, si hubieran buscado cambios epigenéticos, también habrían acabado con las manos vacías.
Los pinos longevos son la excepción del mundo biológico, pero no son únicos desafiando al envejecimiento. El hidrozoo de agua dulce llamado Hydra vulgaris también ha evolucionado para desafiar la senescencia. En las condiciones adecuadas, estos diminutos cnidarios han demostrado un increíble rechazo a envejecer. En libertad, tal vez solo vivan unos pocos meses, sujetos a la depredación, las enfermedades y la desecación. Pero en los laboratorios se han mantenido con vida hasta cuarenta años, sin signos de detenerse ahí, y los indicadores de salud no difieren de forma significativa entre los jovencísimos y los viejísimos.
Hay un par de especies de medusas que son capaces de regenerarse por completo a partir de partes de un cuerpo adulto, y se han ganado el apodo de «medusas inmortales»: la elegante medusa luna, la Aurelia aurita, de la costa Oeste de Estados Unidos, y la Turritopsis dohrnii, de apenas un centímetro de largo, de la costa mediterránea, son capaces de regenerarse, pero supongo que la mayoría de las medusas lo hace. Solo tenemos que observar. Si separas uno de estos increíbles animales en células únicas, estas dan vueltas hasta que forman aglomeraciones que luego se ensamblan hasta formar un organismo completo, como el cíborg T-1000 en Terminator 2, seguramente reseteando el reloj del envejecimiento en el proceso.
Por supuesto, los humanos no queremos que nos desintegren en células únicas para ser inmortales. ¿Qué sentido tiene reensamblarse o generarse si no tienes recuerdos de tu vida actual? Bien podríamos reencarnarnos en su lugar.
Lo importante es lo que estos equivalentes biológicos de Benjamin Button, el personaje de F. Scott Fitzgerald que envejecía al revés, nos enseñan: que el envejecimiento celular se puede resetear por completo, algo que estoy convencido de que lograremos algún día sin perder nuestra sabiduría, nuestros recuerdos ni nuestra alma.
Aunque no es inmortal, el tiburón boreal Somniosus microcephalus sigue siendo un animal impresionante y está mucho más emparentado con nosotros. Es casi del mismo tamaño que un tiburón blanco y no alcanza la madurez sexual hasta los ciento cincuenta años. Los investigadores creen que el océano Ártico podría ser el hogar de los tiburones boreales que nacieron antes de que Colón se perdiera en el Nuevo Mundo. Según la datación por carbono 14, un ejemplar enorme podría haber vivido más de quinientos diez años, al menos hasta que los científicos lo capturaron para poder dictaminar su edad. El hecho de que las células de este tiburón envejezcan o no es un debate científico abierto; muy pocos biólogos habían estudiado al S. microcephalus hasta hace unos años. Como mínimo, este longevo vertebrado experimenta el proceso del envejecimiento muy pero que muy despacio.
En términos evolutivos, todas estas formas de vida están más emparentadas con nosotros que la levadura, y solo hay que pensar en lo que hemos aprendido del envejecimiento humano gracias a ese diminuto hongo. Pero cabe perdonar que, al pensar en la distancia que hay entre los pinos, los hidrozoos, los peces cartilaginosos y los mamíferos como nosotros en el gigantesco árbol de la vida, nos digamos: «No, todas estas cosas son demasiado distintas».
¿Y qué pasa con cualquier otro mamífero? ¿Un primo de sangre caliente productor de leche y creador de vida?
En 2007, unos cazadores aborígenes de Alaska capturaron una ballena boreal a la que, una vez despiezada, le encontraron la cabeza de un viejo arpón clavada en la grasa. Los historiadores determinaron más adelante que el arma se había fabricado a finales del siglo XIX y estimaron la edad de la ballena en unos ciento treinta años. Este hallazgo encendió la chispa de un renovado interés por la Balaena mysticetus, y posteriores investigaciones, gracias a un método para determinar el envejecimiento que mide los niveles de ácido aspártico en el cristalino de los ojos de la ballena, estimaron que aquella ballena boreal tenía doscientos once años cuando la capturaron los balleneros nativos.
Que las ballenas boreales hayan sido seleccionadas para gozar de una longevidad excepcional entre todos los mamíferos tal vez no debería sorprender. Tienen pocos depredadores y pueden permitirse construirse un cuerpo longevo y procrear despacio. Seguramente mantienen su sentido de la supervivencia en alerta constante, reparando las células mientras conservan un epigenoma estable, y asegurándose, por tanto, de que la sinfonía de células toque durante siglos.
¿Pueden estas especies longevas enseñarnos a vivir más sanos y durante más tiempo?
En términos de aspecto y de hábitat, los pinos, las medusas y las ballenas son muy distintos de los humanos. Pero en otros términos nos parecemos mucho. Piensa en las ballenas. Como nosotros, son mamíferos complejos, gregarios, comunicativos y conscientes. Compartimos 12.787 genes conocidos, incluidas algunas variantes interesantes del gen llamado FOXO3. También conocido como DAF-16, este gen se identificó por primera vez como un gen de la longevidad en ascárides por la investigadora de la Universidad de California en San Francisco Cynthia Kenyon. Descubrió que era esencial para los defectos en la ruta de la hormona de la insulina para duplicar la esperanza de vida del gusano. Tiene un papel protagonista en el circuito de supervivencia: el DAF-16 codifica una pequeña proteína transcriptora que se une a la secuencia de ADN TTGTTTAC y funciona con sirtuinas para aumentar la supervivencia celular cuando las cosas se ponen feas.
En los mamíferos hay cuatro genes DAF-16, llamados FOXO1, FOXO3, FOXO4 y FOXO6. Si te da en la nariz que los científicos a veces complicamos las cosas sin necesidad, puede que tengas razón, pero no en este caso. Los genes de una misma «familia genética» han acabado con denominaciones distintas porque se nombraron antes de que se pudieran descifrar con facilidad las secuencias de ADN. Es parecido a cuando, con no poca frecuencia, alguien quiere que le analicen el genoma y descubre que tiene un hermano en la otra punta de la ciudad. DAF-16 es el acrónimo para la formación de larvas dauer. En alemán, Dauer significa «de larga duración», algo relevante para lo que te estoy contando. Resulta que los gusanos se convierten en dauer cuando están hambrientos o agazapados, resguardados hasta que las cosas mejoren. Las mutaciones que activan DAF-16 aumentan la longevidad poniendo en marcha el programa de defensa del gusano incluso cuando las cosas van bien.
La primera vez que me encontré el FOXO/DAF-16 fue en la levadura, donde se conoce como MSN2 (por sus siglas en inglés), que significa «supresor multicopia del regulador epigenético SNF1 (AMPK)». Al igual que el DAF-16, el trabajo de MSN2 en la levadura es activar los genes que alejan las células de la muerte celular y las llevan hasta la resistencia al estrés. Descubrimos que, cuando las calorías están restringidas, MSN2 aumenta la vida de la levadura incrementando los genes que reciclan NAD, dándoles así a las sirtuinas un incentivo.
Ocultos bajo la forma tan retorcida en la que los científicos a veces hablamos de la ciencia se repiten varios temas: sensores de baja energía (SNF1/AMPK); factores de transcripción (MSN2/DAF-16/FOXO); NAD y sirtuinas; resistencia al estrés; y longevidad. No es una coincidencia: todas estas cosas son partes esenciales del antiguo circuito de supervivencia.
Pero ¿qué pasa con los genes FOXO en los seres humanos? Se han encontrado ciertas variantes llamadas FOXO3 en comunidades humanas en las que se sabe que las personas disfrutan de una vida más larga y saludable, como los habitantes de la cuenca del río Rojo, en China. Estas variantes del FOXO3 seguramente activan las defensas del cuerpo contra las enfermedades y el envejecimiento no solo cuando las cosas van mal, sino a lo largo de toda la vida. Si te analizaran el genoma, podrías comprobar si tienes alguna de las variantes del FOXO3 que se asocian a una vida larga. Por ejemplo, tener una variante C en vez de una variante T en la posición rs2764264 se asocia con una vida más larga. Dos de nuestros hijos, Alex y Natalie, heredaron dos C en esta posición, una de Sandra y otra mía, de modo que, al ser los demás genes iguales, y siempre que no adopten un estilo de vida espantoso, deberían de tener más probabilidades de alcanzar los noventa y cinco años que yo, con una única C y una única T, y muchas más que alguien con dos T.
Merece la pena pararse a pensar en lo increíble que es que encontremos básicamente los mismos genes de la longevidad en cada organismo del planeta: plantas, levadura, gusanos, ballenas y humanos. Todas las criaturas vivas proceden del mismo primordio que nosotros. Cuando miramos a través de un microscopio, todos estamos hechos del mismo compuesto. Todos compartimos el circuito de supervivencia, una red de protección celular que nos ayuda cuando las cosas van mal. Esta misma red es nuestra perdición. Los daños severos, como las cadenas rotas de ADN, no se pueden evitar. Sobreexplotan el circuito de supervivencia y cambian la identidad celular. Todos estamos sujetos al ruido epigenético que, según la teoría del envejecimiento por pérdida de información, provoca el envejecimiento.
Sin embargo, diferentes organismos envejecen a diferentes ritmos. Y a veces, según parece, no envejecen en absoluto. ¿Qué le permite a una ballena mantener el circuito de supervivencia sin alterar la sinfonía epigenética? Si el pianista pierde su habilidad, ¿cómo es posible que una medusa restaure la suya?
Son preguntas que me han estado guiando mientras consideraba el rumbo de nuestra investigación. Lo que pueden parecer sueños o ideas sacadas de la ciencia ficción se basan sin lugar a duda en la investigación. Es más, reciben el respaldo de la certeza de que algunos de nuestros parientes cercanos han averiguado la forma de curar el envejecimiento.
Y si ellos pueden, nosotros también.
EL PAISAJE DE NUESTRA VIDA
Antes de que a la mayoría de la gente se le ocurriera siquiera la idea de trazar el mapa de nuestro genoma; antes de que tuviéramos la tecnología necesaria para trazar el mapa del epigenoma completo de una célula y comprendiéramos cómo se compacta el ADN para activar y desactivar los genes, el biólogo del desarrollo Conrad Waddington ya iba más allá.
En 1957, este profesor de genética de la Universidad de Edimburgo intentaba comprender cómo un embrión en fase primera podía transformarse desde un grupo de células indistintas (cada una exacta a la siguiente y con el mismo ADN) hasta los miles de tipos de células que hay en el cuerpo humano. Tal vez no es de extrañar que las inquietudes de Waddington llegaran con los primeros pasos de la revolución digital, al mismo tiempo que Grace Hopper, la madre de la programación computacional, cimentaba el primer lenguaje de programación usado de forma masiva, el COBOL. En resumen, lo que Waddington quería comprobar era cómo las células, que usaban todas el mismo código, producían programas diferentes.
Tenía que haber más que genética en escena: un programa que controlaba el código.
Waddington concibió un «paisaje epigenético», un mapa tridimensional en relieve que representa el mundo dinámico en el que existen nuestros genes. Más de medio siglo después, el paisaje de Waddington sigue siendo una metáfora útil para comprender por qué envejecemos.
En el mapa de Waddington, una célula madre embrionaria se representa como una canica en la cima de una montaña. Durante el desarrollo embrionario, la canica rueda montaña abajo y se detiene en uno de los cientos de valles, que representan un tipo de célula distinto en el cuerpo. Esto se llama «diferenciación». El epigenoma guía a la canica, pero también ejerce gravedad una vez posada la célula, asegurándose de que no vuelva montaña arriba o de que no se traslade a otro valle.
El lugar de descanso final se conoce como «el destino de la célula». Antes creíamos que era un callejón sin salida, un camino irreversible. Pero en biología no hay nada parecido al destino. En la última década, hemos descubierto que las canicas del paisaje de Waddington no están fijas; tienen la terrible tendencia a moverse con el tiempo.
A nivel molecular, lo que sucede con las canicas que ruedan por la ladera de la montaña es que se activan y se desactivan diferentes genes, guiados por los factores de transcripción, las sirtuinas y otras enzimas, como las metiltransferasas de ADN (DNMT) y las metiltransferasas de histonas (HMT), que marcan el ADN y sus proteínas compactadoras con etiquetas químicas que les ordenan a la célula y a sus descendientes que se comporten de cierta manera.
Lo que no se suele apreciar en general, ni siquiera en los círculos científicos, es lo importante que es la estabilidad de esta información para nuestra salud a largo plazo. Verás, la epigenética fue durante mucho tiempo el ámbito de los científicos que estudian los comienzos de la vida, no de la gente como yo, que estudia el extremo opuesto.
Una vez que una canica se asienta en el paisaje de Waddington, suele quedarse allí. Si todo sale bien durante la fertilización, el embrión evoluciona a feto, luego a bebé, luego a niño pequeño, luego a adolescente y luego a adulto. Las cosas suelen marchar bien durante la juventud. Pero el tiempo corre.
Cada vez que hay un ajuste radical en el epigenoma, como, digamos, después de daños en el ADN por el sol o por una radiografía, las canicas se agitan. Imagínate un pequeño terremoto que altera un poquito el mapa. A lo largo del tiempo, tras repetidos terremotos y la erosión de las montañas, las canicas se deslizan por la ladera hacia un nuevo valle. La identidad de una célula cambia. Una célula epitelial empieza a comportarse de forma distinta y activa los genes que en el útero estaban silenciados y que deberían seguir estándolo. Ahora es célula epitelial en un 90 por ciento y otro tipo de célula en un 10 por ciento, con propiedades de neuronas y de células renales. La célula se vuelve una inútil en cosas que deberían hacer las células epiteliales, como crear pelo, mantener la piel elástica y cicatrizar las heridas.
En mi laboratorio decimos que la célula se ha «exdiferenciado».
Todas las células sucumben al ruido epigenético. El tejido formado por miles de células se convierte en una amalgama, una mezcla, un batiburrillo de células.
Como recordarás, el epigenoma es intrínsecamente inestable porque es información analógica (basada en un número infinito de valores posibles) y, por tanto, es difícil evitar la acumulación de ruido y casi imposible de duplicar sin cierta pérdida de información. Los terremotos son una realidad. El paisaje está en constante cambio.
Si el epigenoma hubiera evolucionado para ser digital en vez de analógico, las paredes del valle tendrían una altura vertical de unos 160.000 metros y la gravedad sería fortísima, de modo que las canicas nunca podrían saltar de un valle a otro. Las células nunca perderían su identidad. Si estuviéramos creados de esta forma, nos mantendríamos sanos durante miles de años, tal vez más.
Pero no estamos creados así. La evolución conforma los genomas y los epigenomas lo justo para asegurar la supervivencia necesaria para asegurar a su vez el reemplazo (y tal vez, con algo de suerte, un poquito más), pero no la inmortalidad. De modo que las paredes de nuestros valles tienen poca altura y la gravedad no es tan fuerte. Una ballena que vive cientos de años seguramente ha desarrollado paredes de valles más altas y sus células mantienen la identidad el doble de tiempo que las nuestras. Sin embargo, ni siquiera ellas viven para siempre.
Creo que la culpa la tienen M. superstes y el circuito de supervivencia. El repetido alejamiento de las sirtuinas y de otros factores epigenéticos de los genes a fin de reparar el ADN roto, para después volver a su lugar, si bien es beneficioso a corto plazo, a la postre es lo que nos provoca el envejecimiento. Con el tiempo, los genes equivocados se activan en el momento y en el lugar inapropiados.
Tal como vimos con los ratones ICE, cuando alteras el epigenoma y lo obligas a lidiar con roturas de ADN, introduces ruido, lo que lleva a la erosión del paisaje epigenético. El cuerpo de los ratones se convirtió en quimeras de células despistadas que funcionaban mal.
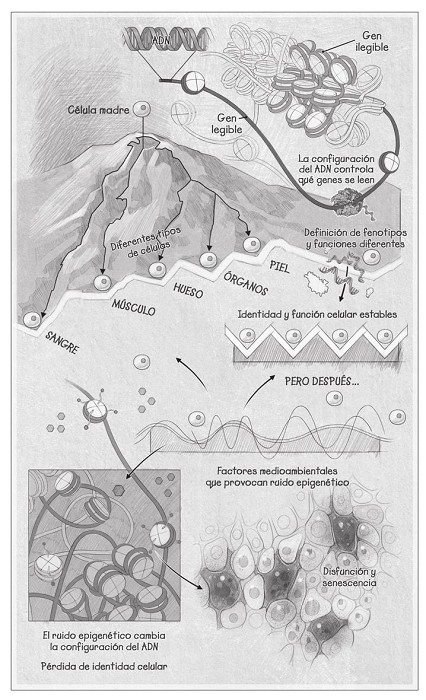
Eso es envejecer. Esta pérdida de información es lo que nos conduce a todos a un mundo de enfermedades coronarias, cáncer, dolor, fragilidad y muerte.
Si la pérdida de información analógica es el motivo único por el que envejecemos, ¿hay algo que podamos hacer al respecto? ¿Podemos estabilizar las canicas manteniendo las paredes de los valles bien altas y una fuerte gravedad?
Sí. Puedo decir con absoluta convicción que podemos hacer algo al respecto.
LA REVERSIÓN SE HACE MAYOR
Hacer ejercicio habitualmente «es un compromiso», dice Benjamin Levine, profesor en la Universidad de Texas. «Pero yo le digo a la gente que piense en el ejercicio como en la higiene personal, como en lavarse los dientes. Debería ser algo que hiciéramos sin pensar para mantenernos sanos.»
No me cabe duda de que tiene razón. La mayoría de las personas haría más ejercicio si ir al gimnasio fuera tan sencillo como lavarse los dientes.
Tal vez algún día lo sea. Los experimentos de mi laboratorio indican que es posible.
—David, tenemos un problema —me dijo una mañana Michael Bonkowski, investigador de posdoctorado, en otoño de 2017, cuando llegué al laboratorio.
No es la mejor forma de empezar el día.
—Vale —repliqué al tiempo que inspiraba hondo y me preparaba para lo peor—. ¿Qué pasa?
—Los ratones —contestó Bonkowski—. No dejan de correr.
Hablaba de unos ratones que tenían alrededor de veinte meses. Eso viene a ser el equivalente de sesenta y cinco años en seres humanos. Les habíamos estado dando una molécula pensada para elevar los niveles de NAD, que creíamos que aumentaría la actividad de las sirtuinas. Si los ratones estaban desarrollando una adicción a correr, sería una buenísima señal.
—Pero ¿cómo va a ser un problema? —le pregunté—. ¡Son noticias estupendas!
—En fin —repuso él—, lo serían de no ser porque han roto la cinta de correr.
Resultaba que el programa para controlar la cinta de correr se había configurado para grabar a un ratón corriendo hasta tres kilómetros. Una vez que los ratones viejos llegaban a esa distancia, la cinta de correr se desconectaba.
—Vamos a tener que empezar el experimento de nuevo —dijo Bonkowski.
Tardé un rato en asimilar esas palabras.
Mil metros es una buena carrera para un ratón. Dos mil metros (unas cinco veces alrededor de una pista de atletismo normal) sería una carrera importante para un ratón joven.
Pero había un motivo por el que habíamos configurado el programa en tres kilómetros: los ratones no corren tanto; sin embargo, estos ratones «ancianos» estaban corriendo ultramaratones.
¿Por qué? Uno de los descubrimientos clave que hicimos, en un estudio publicado en 2018, fue que, cuando los tratábamos con una molécula que elevaba los niveles de NAD que a su vez activaba la enzima SIRT1, las células endoteliales de los ratones ancianos, que recubren los vasos sanguíneos, se introducían en zonas de músculo que no conseguían mucho riego sanguíneo. Se formaban nuevos vasos sanguíneos, capilares, que aportaban el más que necesario oxígeno, eliminaban el ácido láctico y los metabolitos tóxicos de los músculos, y revertían una de las causas más importantes de fragilidad tanto en ratones como en humanos. Por eso, estos ratones viejos se convirtieron de repente en grandes corredores de maratones.
Dado que las sirtuinas se habían activado, el epigenoma de los ratones se estaba estabilizando. Las paredes del valle estaban creciendo. La gravedad cada vez era más fuerte. Y las canicas de Waddington estaban regresando a su lugar correspondiente. El recubrimiento de los capilares respondía como si los ratones estuvieran ejercitados. Era un ejercicio físico mimético, el primero de este tipo, y una señal inequívoca de que algunos aspectos de la «reversión» del envejecimiento eran posibles.
Todavía no conocemos todas las causas de por qué sucede esto. No sabemos qué clase de moléculas funcionan mejor para activar las sirtuinas ni en qué dosis. Se han sintetizado cientos de precursores de NAD distintos y hay ensayos clínicos en activo para responder estas preguntas y muchas más.
Pero eso no quiere decir que tengamos que esperar para aprovechar todo lo que hemos aprendido sobre cómo activar el circuito de supervivencia y vivir más y mejor. No tenemos que esperar para sacarle partido a la teoría del envejecimiento por pérdida de información.
Podemos tomar medidas ahora mismo para vivir mucho más tiempo y mejor. Podemos hacer algo para ralentizar, detener e incluso revertir algunos aspectos del envejecimiento.
Pero, antes de hablar de las medidas que podemos tomar para combatir el envejecimiento; antes de explicar las intervenciones avaladas por la ciencia que prometen cambiar de forma radical nuestra manera de pensar acerca del envejecimiento; antes de hablar siquiera de los tratamientos y de las terapias que cambiarán por completo la historia de nuestra especie, tenemos que responder una pregunta vital.
¿Deberíamos hacerlo?